Charcot-Marie-Tooth Sendromu
CHARCOT-MARIE-TOOTH hastalığı, üç ayrı doktor tarafından tanımlandıktan sonra 1886 yılında bu ismi almıştır. Profesör Jean-Martin Charcot (1825-1893) Pariste birlikte çalıştıkları öğrencisi Pierre Marie (1853-1940) ve Londra’da Dr. Howard Tooth (1926-1956)
İlk olarak ayağı yukarı doğru kaldırmayı sağlayan ve baldırın ön kısmından aşağı inen peroneal kas etkilediğinden CMT aynı zamanda peroneal muskuler atrofi olarak ta anılmaktadır (PMA) . Zayıflamış peroneal kas dağınık yürümeye, düşük ayak takılıp düşmeye neden olduğundan parmak uçları kurtuluncaya kadar hasta bacağını kaldırma gereğini hisseder ve yere koyduğunda ise ayak bir tarafa eğilir.
CMT nin üçüncü ve en son adı HMSN yani (herediter motor and sensoriel neuropathy) kalıtsal motor ve duyu siniri bozulmasıdır. Bu isim, sendromu daha eksiksiz tanımlamaktadır çünkü CMT kalıtsaldır, hem hareket ve hem de duyu sinirlerini etkileyebilmektedir. Çoraplarının üzerinden bacak veya ayaklarıyla hissedemeyen, ayak bilek ve parmaklarını oynatamayan ve hatta dizden alt tarafını hiç hareket ettiremediği gibi hiçbir şey hissetmeyen hastalar vardır. En çok görülen de hareket kaybıdır.
CMT, sinir üzerindeki myelin veya miyelin izolasyon kılıfının bütünlüğünü koruyamayıp bozulması ve buna bağlı olarak beyinden alınan mesajların sinirler vasıtasıyla kaslara düzenli iletilememesi nedeniyle ortaya çıkan, primer bir sinir hastalığıdır. Bu durum da doğuştan kasları normal yapıda olan CMT hastalarını muskuler distrofisi olanlardan ayırt edici bir niteliktir. Kas atrofisi CMT’nin sinirleri etkilemesinden ve beyinden gelen hareket mesajlarının düzenli iletilmemesinden kaynaklanır. Bu nedenle, kullanılabiliyor olsa da kaslar atrofiye olmaktadır.
Muskuler distrofisi olanların ise doğuştan kaslarıyla ilgili sorunları vardır.
İsim olarak fazla tanınmamasına rağmen CMT nadir görülen bir hastalık değildir. Aile içinde nesilden nesile taşınmaktadır. Doğru teşhis konan bir aile üyesi, ailedeki herkes için bir ışık teşkil etmektedir. Ailede CMT tanısı konmuş bir birey varsa sonraki kuşak bireylerinde bu hastalığın görülme muhtemellığı yüksektir.
CMT kalıtım yoluyla 3 şekilde geçmektedir fakat olayların çoğunda otozomal dominant örnekler vardır bu da ebeveynden çocuğa doğrudan geçişi ifade eder. Bu kalıtım formunda her hamilelikte, çocuğun CMT hastası olma şansı %50 dir.
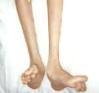
Dıştan görünen belirtiler doktorların CMT tanısına başlamak için aradıkları özelliklerdir. CMT nin ilk belirtileri; dizden aşağı bacağa ince bir görüntü verecek şekilde baldır bölgesindeki adalenin zayıflaması, düşük ayakla yürüme, yüksek köprülü veya düz tabanlı ayaklar ile diğer kemik deformiteleri ibik veya çekiç ayak parmağı bilek zayıflığı ayak bilekte duyu ve hareket kaybıdır. İnsanlar çoğu zaman hantallaştıklarından dolayı bir şeye takılıp düştüklerini zannederler ama bu anda aslında CMT tecrübesi geçirmektedirler. CMT li ayaklar çok normal bir görüntü de verebilir.
Üst ekstremitelerdeki belirtiler, parmaklar, el ve bilekte ona bağlı olarak kavramada zayıflık, kas zayıflamasına bağlı olarak baş parmak hareketlerinin azalması, el ve bilekte duyu ve hareket kaybı şeklindedir. İnsanlar çoğu zaman dikkatsiz ve savruk zanneder çünkü ellerinden bir şeyleri düşürürler veya düğme, para ve toplu iğne gibi eşyayı yerden toplayamazlar. Normal bir görünümde olmasına rağmen CMT elleri etkiler ve zayıflatır.
Ayak kaslarının zayıflamasından denge her zaman için etkilenir ve yürüme anında hız değiştirmeyi veya aniden durmayı kompanse edemez. CMT li bir hasta için ayakta sabit durmak çok zordur bunu sağlamak için bir şeye dokunmak tutunmak ihtiyacını hisseder. Reflekslerin zayıflığı veya yokluğu yanında şüphesiz ailenin hikayesini bilmesi hekime CMT teşhisi koymada çok yardımcı olur .
Her CMT hastası için bir tecrübe olarak görünen yorgunluk, bitkinlik en önemli semptomlardan biridir. Bu konuda ağır ve ılımlı bir yürüyüş önerilmekte ve iyi sonuçlar alınmaktadır. Erken yaşlarda CMT belirtileri gösterenlerde skoliosis ve diğer spinal deformiteler de oluşabilir ve baz hastaları deforme kalça oyukları ile dünyaya gelirken, bazı CMT hastalarında kalça ve diz çıkıkları yaşamının bir parçasıdır.
CMT, en kesin şekilde EMG (elektromyogram) uygulanarak teşhis edilir. EMG kasların irritabilite ve fonksiyonlarını ölçüp, sinir hücrelerinin uyarı taşıma hızını test ve sinirlerin uyarıları gönderme ve alma yeteneklerini tespit eden bir cihazdır.
Birçok örneklerinde CMT kalıtsal karakter gösterir. Babanın veya annenin CMT hastası olması haline göre ailedeki bireylerin şansı önceliklidir. En çok görülen A1 tipi CMT niz varsa bunu doğrudan anne veya babanızdan kalıtım yoluyla almanız güçlü bir ihtimaldir. Ebeveyniniz bu hastalığı çok hafif geçirmiş hatta hasta olduğu dahi anlaşılmamış olabilir. Her hamilelik için çocuğun CMT tip A1 i kalıtsal olarak alma şansı %50 dir. CMT aynı zamanda resesif ve x genine bağlı olarak taşınıp aktarılabilmekte veya ailede hiç CMT hastası olmasa da kendiliğinden oluşan bir mutasyondan sonra ortaya çıkabilmektedir. Bu durumda ailede CMT yi taşıyan ve aktaran ilk kişi olabilmektesiniz. Genetik bir aktarma mı yoksa mutasyonel mi olduğu noktası yapılacak kan ve DNA testleriyle açıklığa kavuşmaktadır. CMT nin en çok görülen tipine, 17 no. lu kromozom üzerindeki periferal myelin protein geninin duplikasyonu sebep olmakta bir çift yerine 3 gen ortaya çıkmaktadır. Kalıtsal nöropatiye neden olan bu genin yok edilmesi CMT nin birçok semptomlarını taşıyan bir hastalık olan ağır felç ve ilave olarak kol veya bacağınız üzerinde eğildiğinizde basınç uygulanan mahallerde çok koyu bir hissizliğe yol açmaktadır.
CMT INT-Canada yayınlarından derlenmiştir.
Siz de fikrinizi belirtin